
Glycosylation is a protein post-translational modification (PTM) that generates extreme proteomic diversity and contributes to a plethora of cellular functions. Protein glycosylation can have an enormous variety of biological consequences, reflecting the molecular diversity encoded in glycan structures. This same structural diversity has imposed major challenges on the development of methods to study the intact glycoproteome.
While there has been much interest in profiling the intact glycoproteome, the complexity of glycoproteoforms (and, more broadly, all proteoforms) remains challenging to completely define. Mass spectrometry (MS) is commonly employed for characterization of complex proteomic samples. A popular strategy for protein identification is the bottom-up shotgun proteomics approach. In this method, a mixture of proteins is subjected to proteolytic digestion, the resulting peptides are separated by LC and detected by MS, and their parent proteins are inferred from the assigned peptide sequences. To convert MS data acquired from proteolytic digests into protein identifications, tandem MS can be used to obtain sequence information for individual peptides, followed by comparison to an in silico proteolytic digest of an organism’s proteome. Typically, only the most abundant peptides are selected for fragmentation, whereas data for those peptides in relatively low quantities are not obtained. An inherent problem in shotgun proteomics is identifying proteins of low abundance, such as biomarkers for disease states, against a background of proteins whose concentrations can span up to 12 orders of magnitude.
To address the unique challenges of global characterization of the intact glycoproteome, a mass-independent chemical glycoproteomics platform, termed “isotope targeted glycoproteomics” or IsoTag was developed by the Carolyn Bertozzi group. The platform comprises four central components: (i) metabolic labeling with a chemically functionalized glycan, (ii) chemical tagging and enrichment using an isotopic recoding affinity probe, (iii) directed tandem MS, and (iv) targeted glycopeptide assignment.
In traditional proteomics approaches, tandem MS is performed on the most abundant species in the full-scan mass spectra to the exclusion of lower-abundance species (Figure 1). In contrast, IsoTaG enables mass-independent, targeted glycoproteomics (Figure 1). Glycopeptides displaying the isotopic signature are computationally detected by the pattern-searching algorithm [1], which produces an inclusion list of m/z values and retention times (±2-min window) for ions bearing isotopically recoded envelopes. Femtomole quantities of tagged proteins at a signal-to-noise ratio of 2.5:1 are detectable with the pattern-recognition algorithm [1]. Owing to the versatility of the algorithm [1], species up to 12 kDa that have incorporated multiple tags are also directed to the inclusion list and have an equal chance of selection for tandem MS.
[1] Palaniappan, K.K., et al. (2011) Isotopic signature transfer and mass pattern prediction (IsoStamp): an enabling technique for chemically-directed proteomics. ACS Chem. Biol. 6 829–36.
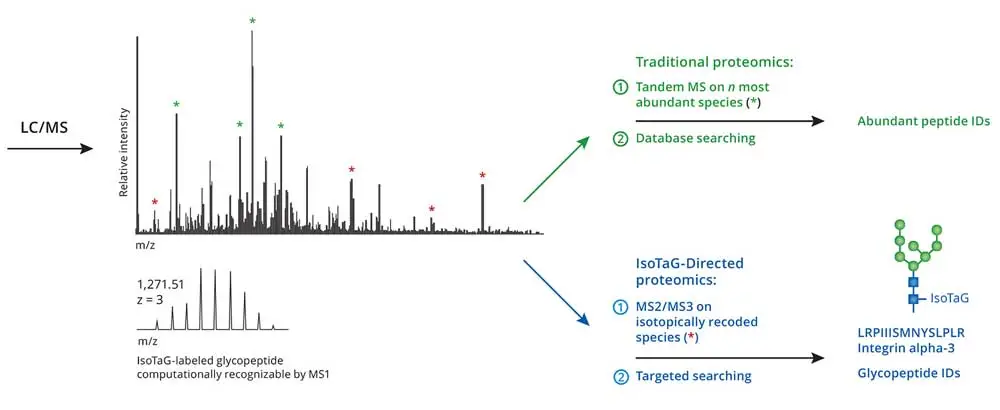
Figure 1. Traditional proteomics and Iso-Tag-directed proteomics workflow
IsoTaG is performed first by the isotopic recoding and enrichment of metabolically labeled glycoproteins followed by directed tandem MS (MS2 or MSn) analysis and intact glycopeptide assignment. Isotopic recoding is accomplished by metabolic labeling of cell or tissue samples with azide- or alkyne-functionalized sugars, followed by chemical conjugation with a biotin probe bearing a unique isotopic signature. Some examples of sugar labels include peracetylated N azidoacetylmannosamine (Ac4ManNAz), which is converted to the corresponding azidosialic acid (SiaNAz), and peracetylated N-azidoacetylgalactosamine (Ac4GalNAz), which is metabolized to label glycans possessing N-acetylglucosamine (GlcNAc) or N-acetylgalactosamine (GalNAc) (not provided with kit).
In order to perform isotopic tagging, the kit provides two cleavable IsoTaG probes encoded by zero [M] and two [M + 2] deuterium atoms. Probes with different encoding can be provided by Click Chemistry Tools though custom synthesis. The IsoTaG probes with zero and two deuterium atoms [M, M + 2] can be used in different proportions: 1:1, 1:2, 1:3 and 1:4. Pattern recognition with an isotopic ratio of 1:3 showed the highest fidelity.
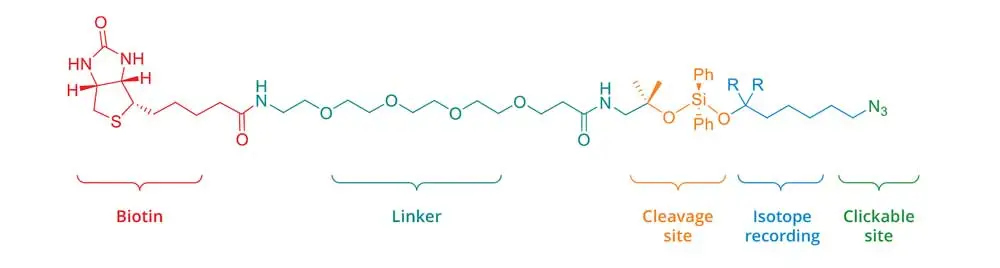
Figure 2. Cleavable IsoTaG probe encoded by zero deuterium atoms [M] (R = H) and two deuterium atoms [M+2] (R = D)
The isotopic signature serves as a computationally recognizable full-scan MS reporter. A computational algorithm, termed isotopic signature transfer and mass pattern prediction – IsoStamp, for the detection of recoded species in full-scan mass spectra was also developed by the Carolyn Bertozzi group. IsoStamp compares observed and predicted isotopic envelopes to identify chemically tagged species in full-scan mass spectra.
IsoTag has the potential to enhance any proteomics platform that employs chemical labeling for targeted protein identification, including isotope-coded affinity tagging, isobaric tagging for relative and absolute quantitation, and chemical tagging strategies for posttranslational modification.
Kit Contents
| Component | Concentration | Amount | Storage | Stability |
|---|---|---|---|---|
| Streptavidin Agarose Resin (Component A) | 50% slurry | 2.5 mL | 4°C | Stable for at least 12 months when stored as directed. |
| DADPS H2 Biotin Azide [M] (Component B) | — | 1 vial | 4°C | |
| DADPS D2 Biotin Azide [M+2] (Component C) | — | 1 vial | 4°C | |
| Copper (II) Sulfate + Protectant (Component D) | 100 mM | 250 μL | 4°C | |
| Reducing Agent (Component E) | — | 100 mg | 4-30°C | |
| Alkyne Labeled BSA (Component F) | — | 0.5 mg | 4°C | |
| 5% Formic acid (Component G) | 5% | 3 mL | 4-30°C |
Materials Required for Enrichment but Not Provided for Capturing of Alkyne-tagged Proteins
- 5-20 mg of cell or tissue extract containing alkyne-modified biomolecules
- Protease Inhibitors (e.g., Sigma P8340)
- High-speed microcentrifuge
- Unlabeled cells or tissue containing the same relative amount of protein (negative control)
- 1.5 ml microfuge tubes
- 1% SDS in 50 mM Tris-HCl
- Solvents: methanol, DI water
- Probe sonicator or endonuclease such as Benzonase®
Materials Required but Not Provided for On-resin Digestion with Protease.
- DTT
- Iodoacetamide
- 8M Urea/100 mM Tris, pH 8
- Acetonitrile
- Mass-spec grade trypsin
- 0.1% TFA
- C-18 desalting cartridges
- Digestion Buffer
- Heat block
- Vacuum Concentrator
Additional information
- The concentration of an azide biotinylation reagent in a click labeling reaction may range from 2 μM to 60 μM. Concentrations below or above this range are also possible, and should be optimized for the specific application. We recommend starting with a 40 μM concentration of the azide reagent, and titrating this amount down in the case of high background or up in the case of low reaction efficiency. The kit provides sufficient amount of the azide reagents to perform 25 labeling reactions using up to 100 μM biotin azide concentration.
- The isotopic ratio (H2:D2 probes or [M]:[M+2] ratio) is application dependent. Pattern recognition with an isotopic ratio of 1:3 showed the highest fidelity. However, investigators may wish to determine the optimal ratio for each application.
- Caution: copper (II) sulfate solution is harmful to aquatic organisms and can cause damage to aquatic environments. Avoid release into the environment. Refer to MSDS.
Material Preparation
| Streptavidin Agarose Resin (Component A) | Ready to use. Stable for 1 year when stored at 4°C. |
| DADPS Biotin Azides (Component B and C) | Dissolve both DADPS Biotin Azide reagents in 1 mL of DMSO. After use, store unused detection reagent stock at -20°C for up to 1 year. Mix H2 and D2 DADPS Biotin Azide probes at desired ratio. Isotopic ratio of 1:3 (100 μL of H2 and 300 μL of D2) can be used for initial experiments. |
| Copper (II) Sulfate (Component D) | Ready to use. Stable for 1 year when stored at 4°C. |
| Reducing Agent (Component E) | Prepare only as much of Reducing Agent (Component E) solution as necessary for that day’s experiment and use on the same day. Weigh 20 mg of Reducing Agent (Component E), add 100 μL of deionized water, vortex until completely dissolved. |
| Positive Control Alkyne Labeled BSA (Component F) | Reconstitute lyophilized BSA Alkyne in 500 μL of 0.5x PBS to obtain 1 mg/ml solution. Add 10 μL BSA Alkyne (Component F) to 1000 μL of unlabeled cell lysate with proteins concentration of 1 mg/mL. This mixture will contain 1% of BSA Alkyne. The amount of BSA Alkyne in the positive control is application dependent and can be adjusted to more closely mimic the expected amount of the alkyne labeled proteins in cell lysate. |
| 5% Formic Acid (Component G) | Ready to use. Stable for 1 year when stored at 4-30°C. |
1. Metabolic Labeling
This protocol details the preparation of metabolically labeled cell lysates for IsoTaG enrichment. The following is a standard protocol for preparation of azido- or alkynyl-sugar labeled cell lysates and conditioned media. However, investigators may wish to determine the optimal concentration of metabolic reagent as well as labeling time individually for each cell type on a small-scale first. Metabolic labeling is critical for successful glycoproteomics and should be carefully assessed for each cell line of interest prior to MS analysis. Metabolic labeling of glycoproteins is dependent on three considerations: first, the sugar precursor selected for labeling; second, access of the sugar precursor to the cell line; and third, glycosylation pathways activated in the cell line. Options for sugar precursor selection include Ac4ManNAz, Ac4GalNAz, Ac4GlcNAz, and Ac4ManNAl, among others. Ac4ManNAz and Ac4ManNAl are reporters of sialic acid. Ac4GalNAz and Ac4GlcNAz are able to access an intracellular epimerase pathway and are incorporated into O-GalNAc, O-GlcNAc, and N-GlcNAc sites.
The efficiency of metabolic labeling is dependent on access of the sugar precursor to the cell line, and testing with several sugar precursors may be necessary for best results. Finally, sugars with access to multiple metabolic endpoints (e.g., Ac4GalNAz) will be incorporated to a set of glycoproteins that are reflective of the glycosylation pathways currently activated in the cell line. In general, human cell lines are permissive to the sugar precursor and cell counts of 107–108 (equivalent to 1–3 mg of total protein) are sufficient inputs for this protocol. In the event that metabolic labeling is prohibitive, alternate options to install bioorthogonal handles to the sample of interest may be considered (e.g., enzymatic labeling). The kit provides alkyne labeled BSA as a positive control.
- Prepare a stock 500 mM solution of metabolic labeling reagent in DMSO to make a 1000X to 5,000X stock solution. Aliquot and store any unused reagent at –20°C. When stored as directed, this stock solution is stable for up to 1 year.
- Seed the appropriate number of cells so they are fully confluent after a 4-day incubation period. To the media, add metabolic labeling reagent (50-100 µM) or DMSO vehicle control containing Ac4GalNAc. Incubate the cells at 37°C for 48 hours in a humidified, 5% CO2 incubator. This procedure yields 3-5 mg of protein.
Note: for analysis of cellular and membrane incorporation only, the current incubation period is sufficient and the user may skip to Preparation of Cell Lysates. - Aspirate the media and wash cells with PBS. Then, change the media to the one containing 100 μM glycan metabolite without FBS additive and incubate cells at 37°C, 5% CO2 for an additional 48 hours.
2. Preparation of Conditioned Media (Optional)
- Transfer the conditioned media to centrifuge tubes.
- Clear the conditioned media by centrifugation (150 g, 3 min).
- Repetitively spin concentrate the clarified media to a final volume of 500 μL (Amicon spin filter, 3700 g, 15–30 min per spin, approximately five spins required). Discard the flow through appropriately.
- Wash the concentrated proteins with 0.1% triton X-100 in PBS (3 × 15 mL) by centrifugal filtration (3700 g, 15–30 min) to remove excess labeling reagent.
- Transfer the residue to a 2 mL microcentrifuge tube. The concentrated conditioned media may be stored for at least 12 months at –80°C.
3. Harvesting Suspension Cells
- Pellet the cells by centrifugation at 400 g for 5 minutes. Discard the supernatant.
- Resuspend the cell pellet in PBS by gently pipetting up and down using 5 mL PBS for cells from a 100 mm dish or 1 mL PBS per well for cells from a 6-well plate.
- Pellet the cells by centrifugation at 400 g for 5 minutes. Discard the supernatant.
- Repeat the PBS wash 2 more times (steps 3.2–3.3) for a total of 3 washes to remove serum.
- Pellet the cells by centrifugation at 400 g for 5 minutes. Discard the supernatant. The cell pellet can be used directly in step 5.1 or flash frozen and stored at –80°C until use.
- For Suspension Cells, pellet them by centrifugation at 400 g for 5 minutes, wash 3 times with PBS, discard supernatant, and use directly in step 5.1 or flash freeze and store the pellet at –80°C.
4. Harvesting Adherent Cells
Note: If analyzing cell surface proteins, do not use trypsin to detach cells, because trypsin cleaves cell surface proteins. The cells can be lysed directly in the culture dish, or, if desired, use a non-enzymatic dissociation buffer or a cell scraper, and pellet the cells. If not used immediately in step 5.1 the cell lysate can be frozen and store it at –80°C until use.
- Cells can be harvested with cell detaching solution (EDTA-free trypsin or non-enzymatic) or proceed immediately to the cell lyse step for preparing samples for the click reaction.
- After removing the medium or cell detaching solution, wash the cells three times with PBS and discard supernatant.
5. Preparation of the Cell Lysates
Do not use DTT, TCEP, or β-mercaptoethanol because they will reduce the azide. Do not use EDTA or any other chelators because they will inhibit the click reaction.
The following is a standard protocol for preparation of cell lysates. However, investigators may wish to determine the optimal protocol (e.g. lysis protocol with cellular fractionation) individually for target proteins localization [2]. Cell lysate preparation is critical for successful glycoproteomics and should be carefully assessed for each target protein subset prior to MS analysis.
- Prepare the lysis buffer by adding protease and phosphatase inhibitors at appropriate concentrations to 1% SDS in 50 mM Tris-HCl, pH 8.0. Alternate lysis protocols (e.g., RIPA buffer, high-salt extraction) are compatible with downstream enrichment.
Note: Protease and phosphatase inhibitors are optional but recommended to ensure sample integrity. - For adherent cells, add 500 μL lysis buffer per 100 mm plate or 200 μL lysis buffer per well of a 6-well plate to the labeled cells. If adding the lysis buffer directly to the plate, tap or rotate the plates so the lysis buffer covers the bottom surface of the plate.
For suspension cell pellet, add 50 μL lysis buffer per 1 × 106 cells. - Incubate the cells for 15–30 minutes on ice, and then tilt the plates and pipet the lysate into a 1.5 mL microcentrifuge tube. If the lysis buffer does not contain Benzonase® endonuclease, the lysate may be very viscous due to the DNA from the lysed cells.
If using Benzonase® endonuclease, proceed to step 5.5. - Sonicate the lysate with a probe sonicator to solubilize the proteins and disperse the DNA.
- Gently vortex the lysate for 5 minutes.
- Centrifuge for 10 minutes at 13,000-20,000 g at 4°C.
- Transfer the supernatant to a clean tube and label as the “soluble fraction”.
- Measure the protein concentration of the three fractions (conditioned media, soluble, and insoluble) by BCA or any other assay, and normalize protein concentration to 5 mg/mL.
If the protein concentration is lower than 5 mg/mL, normalize to the lower value and adjust the volumes used in following steps accordingly. - The protein sample is now ready for the click labeling reaction with IsoTag Biotin Probes.
[2] Baghirova S., et al. (2015). Sequential fractionation and isolation of subcellular proteins from tissue or cultured cells. MethodsX. 2:440-15.
6. Click Labeling Reaction with IsoTag Biotin Probe
This protocol provides guidelines for the click reaction using 600 μL of cell lysate. However, it can be suited for smaller or larger volumes with adjustments of volumes up or down accordingly. The concentration of biotin azide reagent in this protocol is set to 40 μM. The concentrations of azide reagent may range from 2 μM to 60 μM; concentrations below or above this range are also possible, and should be optimized for each sample type. If a higher concentration of biotin probe required, increase only the volume of IsoTag reagent in the click labeling reaction leaving all other amounts unchanged.
- For each alkyne-modified protein lysate sample, add the following to a 2.0 mL microfuge tube, then vortex briefly to mix:
• 20 µL IsoTag Reagent (Component B/C, mixed at desired ratio).
• 50 µL Copper (II) Sulfate Mix (Component D).
• 30 µL Reducing Agent. - Vortex the tube briefly to mix. This pale blue solution turns colorless after the addition of the Reducing Agent.
- Add the solution from Step 6.2 to 600 μL of cell lysate.
- Vortex continuously or rotate end-over-end for one hours at room temperature.
- Add the labeling reaction to 3 mL of cold (-20°C) methanol, 0.75 ml of Chloroform and 2.1 mL of water. Cool it to -20°C for 1 hour.
Note: cold (-20°C) acetone can be used in place of methanol:chloroform:water mixture. - Centrifuge for 10 minutes at 13,000-20,000 g, carefully remove upper aqueous layer without disturbing interface layer containing proteins.
- Add 450 µL of methanol, vortex briefly.
- Centrifuge for 5 minutes at 13,000-20,000 x g to pellet protein. Carefully remove and discard supernatant.
- Open the lid to microfuge tube and allow protein pellet to air dry. Do not overdry the pellet!
7. Binding Biotinylated Proteins to Streptavidin Agarose Resin
- Resuspend air-dried protein pellets (from Step 6.9) by bath sonication in 800 μL of resuspension buffer (50 mM Tris, 150 mM NaCl and 1% SDS). Other denaturing pull down buffers (for example 6 M urea, 2 M thiourea, and 10 mM HEPES) are also compatible with downstream analysis.
- Solubilize the proteins by brief sonication with a probe tip sonicator. Save an aliquot (50 μL, “input”) for analysis by Western blot. A clear solution will result, indicating a fully solubilized pellet.
- Wash the streptavidin agarose resin (200 μL of slurry per sample) with PBS (2 × 1 mL) and with resuspension buffer (2 x 1 mL) by either vacuum filtration or centrifugal force (2000 g, 3 min).
- Resuspend the slurry with resuspension buffer (200 μL) and add the slurry to the solubilized proteins.
If Western blot analysis indicates incomplete capture of biotinylated proteins, increase slurry amount in future. - Incubate the resulting mixture for 2 hours at ambient temperature with end-over-end rotation.
- Pellet the beads by centrifugation (2000 g, 3 min).
- Separate the supernatant. Save an aliquot (100 μL, “flow-through”) for analysis by Western Blot.
- Wash the beads with resuspension buffer (1 x 1 mL), 1% SDS in PBS (2 × 1 mL), and PBS (2 x 1mL) in succession. Pellet the beads by centrifugation (2000 g, 3 min) between washes, and discard the wash.
Note: Stringency of washing may be increased (e.g., 8 M urea washes) if high protein background is observed by MS or Western blot. - Resuspend the washed beads in PBS (200 μL). Save an aliquot (10 μL, “capture”) of the slurry for analysis by Western blot.
8. On Bead Trypsin Digestion of Enriched Glycoproteins
- Add to the resin 10 μL of 1 M DTT, vortex briefly.
- Heat to 70°C on a heat block for 15 minutes, and then cool to room temperature for 15-30 minutes.
- Centrifuge resin for 5 minutes at 2000 g, aspirate and discharge the supernatant taking care not to aspirate the resin.
- Add 1 mL of 40 mM iodoacetamide solution to the resin, vortex to resuspend the resin, incubate the reaction in the dark for 30 minutes at room temperature.
- Pellet the beads by centrifugation (2000 g, 3 min) and wash once with PBS (1 mL).
- Cap the bottom of the spin column, then add 300 μL of digestion buffer (100 mM Tris, 2 mM CaCl2, 0.5 M urea) to the resin.
- Add 3 μg of trypsin from a stock solution to the resin slurry. Gently mix the slurry, then incubate at 37°C for 6-12 hours with over-the-end rotation.
Note: Alternative proteases and trypsin digestion procedures are also compatible with this protocol and may be used to increase glycopeptide coverage. - Pellet the beads by centrifugation (2000 g, 3 min), and collect the supernatant digest.
- Wash the beads with PBS (1 x 200 μL) and water (2 × 200 μL).
- Combine the washes with the supernatant digest to form the “trypsin fraction”.
- Concentrate the “trypsin fraction” to dryness using a speedvac set to 40°C.
- The “trypsin fraction” may be stored for at least 12 months at –80°C.
9. Cleavage and Recovery of Glycopeptides
- Cleave the probe by adding of 5% formic acid–water (200 μL) to the beads and incubate for 30 min at ambient temperature with end-over-end rotation.
- Pellet the beads by centrifugation (2000 g, 3 min), and collect the supernatant.
- Repeat Step 9.1 a second time.
- Wash the beads with 50% acetonitrile–water + 1% formic acid (2 × 400 μL).
- Combine the washes with the cleaved supernatant to form the “cleavage fraction”.
- Concentrate the “cleavage fraction” to dryness using a Speedvac heated to 40°C. Store at –20°C until desalting and MS analysis.
- For long term storage, the “cleavage fraction” may be stored for at least 12 months at –80°C.
10. Preparation of Digest for Mass Spectrometry Analysis
The following protocol is provided for desalting using ZipTip C18 P10. Sample recovery for typical peptides is usually > 85%, but could be as low as 35% for hydrophilic peptides. Other protocols for sample preparations for MS also can be used.
- Resuspend the dried “trypsin fraction” and “cleavage fraction” in 1% formic acid in water (25.0 μL). Gently vortex the sample and briefly centrifuge to collect all liquid at the bottom.
- Wet the tip by aspirating 10 μL of 50% ACN in water and then discarding solvent. Repeat once.
- Equilibrate tip by aspirating 10 μL of 0.1% aqueous formic acid and discarding solvent. Repeat twice.
- Load the peptides by pipetting the sample 10–15 times. Carefully pipette the sample such that air bubbles do not pass through the C18 tip. Air bubbles decrease loading capacity.
- Wash the pipette tip with 1% formic acid in water (50 μL) and discard the eluent.
- Slowly aspirate 25μL of 50% ACN:Water with 1% formic acid. Repeat twice.
- Concentrate the sample using a speedvac heated to 40°C. Store at –20°C until analysis by LC/MS. For long-term storage, the desalted samples may be stored for at least 12 months at –80°C.
Selected References:
Woo, C.M., et al. (2015). Isotope-targeted glycoproteomics (IsoTaG): a mass-independent platform for intact N- and O-glycopeptide discovery and analysis. Nat Methods., 12: 561−7.
Woo, C. M., et al. (2017). Development of IsoTaG, a Chemical Glycoproteomics Technique for Profiling Intact N- and O‑Glycopeptides from Whole Cell Proteomess. J. Proteome Res., 16: 1706−18.
Gao, G., et al. (2017). Small Molecule Interactome Mapping by Photoaffinity Labeling Reveals Binding Site Hotspots for the NSAIDs. J. Am. Chem. Soc., 140: 4259−68.
Weerapana, E., et al. (2010). Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature, 648: 790−5.
Woo, C.M.., et al. (2017). Mapping and Quantification of Over 2000 O-linked Glycopeptides in Activated Human T Cells with Isotope-Targeted Glycoproteomics (Isotag). Mol. Cell.Proteomics., 17: 764−75.
Troubleshooting
| Problem | Possible Cause | Solution |
|---|---|---|
| Low yield of enriched peptides | Inefficient protein click labeling with biotin or low abundance of alkyne-tagged proteins | Increase lysate concentration (use more cells) or pre-enrich the proteins (e.g. soluble lysate, membrane lysate, lectin enrichment, etc.). Confirm peptide recovery by measuring A280 after digestion |
| Inefficient digestion of resin-bound proteins | Use high quality trypsin | |
| High background with unlabeled control cells | Insufficient washing of resin | Increase column washes Use only high purity reagents Prepare filtered buffers fresh Ensure proper preparation of copper catalyst solution |
| Signal suppression during MS analysis | SDS contamination in digest | Wash the resin thoroughly after the SDS wash with another buffer such as 8M urea and 20% acetonitrile to remove all traces of SDS detergent |
